MetaboAnalystはWebでメタボロミクス解析が可能なサイトです。そして、MetaboAnalystの解析の大部分はRのパッケージによっておこなわれています。
MetaboAnalyst:https://www.metaboanalyst.ca/home.xhtml
しかし、組織のセキュリティ規則でWebへデータをアップロードできない場合もあるかと思います。
そのため、MetaboAnalystのサイトではWebではなく、ローカル環境で同様の解析が実施できる「MetaboAnalystR」パッケージのインストール方法が記述されています。
しかし、記述されている構築方法ではいくつかのインストールでエラーが起こります。調べたところ解決方法の情報がすくなく簡単ではありません。
そのため、MetaboAnalystをローカル環境で利用したくとも、導入できず苦労している方もいると思います。
そこで、「MetaboAnalystR」パッケージの構築方法を紹介します。MetaboAnalystでの解析方法が決まっているのであれば、MetaboAnalystで出力されるRコマンドさえ手に入れるとルーチンで実行できるので大変便利だと思います。
実行コマンドは新規にR version 4.3.1をインストールした環境で「MetaboAnalystR」パッケージのバージョン 4.0.0を導入できることを確認しています。
事前準備
「MetaboAnalystR」パッケージのインストールに必要な準備です。Rのバージョンに合わせて紹介するリンクからRToolsをダウンロードしインストールしてください。
RTools:https://cran.r-project.org/bin/windows/Rtools/
次に下記リンクから「SSPA_2.22.1.tar.gz」をデスクトップにダウンロードします。ダウンロードはページ下部の「Source Pakage」横のリンクをクリックしてください。
SSPA_2.22.1:https://www.bioconductor.org/packages/3.8/bioc/html/SSPA.html
最後に下記コマンドをRで実行します。コメントアウト部分は必要に応じて実行してください。
# Rのバージョンに合わせてRToolsをダウンロードして実行
# https://cran.r-project.org/bin/windows/Rtools/
# devtoolsパッケージのインストール
# install.packages("devtools", type = "source")
# 上手くいかない場合は上記コマンドで再度インストールする
install.packages("devtools")
# BiocManagerパッケージのインストール
install.packages("BiocManager")
# ellipseパッケージをインストール
install.packages("ellipse")
# rjsonパッケージをインストール
install.packages("rjson")
# OptiLCMSパッケージをインストール
devtools::install_github("xia-lab/OptiLCMS", build = TRUE, build_vignettes = FALSE, build_manual =TRUE)
## 以下場合によっては必要かもしれません
# rlangのインストール:Ver1.0.5以上が必要
# install.packages("rlang")
# cliのインストール:Ver3.3.0以上が必要
# install.packages("cli")
##
# 以下からSSPA_2.22.1.tar.gzをダウンロード
# https://www.bioconductor.org/packages/3.8/bioc/html/SSPA.html
# SSPAに必要パッケージのインストール
BiocManager::install(c("qvalue", "limma"), force = TRUE)
# 下記コマンドを実行しSSPA_2.22.1.tar.gzを選択してインストール
install.packages(file.choose(), repos = NULL, type = "source")「MetaboAanalyst」パッケージに必要なパッケージをインストール
下記コマンドをRで実行してください。
# 準備
metanr_packages <- function(){
metr_pkgs <- c("impute", "pcaMethods", "globaltest", "GlobalAncova", "Rgraphviz", "preprocessCore", "genefilter", "sva", "limma", "KEGGgraph", "siggenes","BiocParallel", "MSnbase", "multtest", "RBGL", "edgeR", "fgsea", "devtools", "crmn")
list_installed <- installed.packages()
new_pkgs <- subset(metr_pkgs, !(metr_pkgs %in% list_installed[, "Package"]))
if(length(new_pkgs)!=0){if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(new_pkgs)
print(c(new_pkgs, " packages added..."))
}
if((length(new_pkgs)<1)){
print("No new packages added...")
}
}
# インストール
metanr_packages()「MetaboAanalyst」パッケージをインストール
下記コマンドを実行してください。
# インストールコマンド
devtools::install_github("xia-lab/MetaboAnalystR",
build = TRUE, build_vignettes = FALSE,
# マニュアルが必要な場合は下記オプションをTRUE
build_manual = FALSE)正しく「MetaboAanalyst」パッケージがインストールされていると、パッケージ読み込み時にメッセージが紹介されます。
# パッケージの読み込み
library("MetaboAnalystR")
MetaboAnalystR 4.0.0 initialized Successfully !
# https://github.com/xia-lab/MetaboAnalystR実行コマンド
MetaboAnalystサイトのData Formatから「Analysis Path」、「Statistics [one factor]
and Biomarker Analysis」、「Concentration table」の「cow_diet.csv」データを使用した解析例です。
Data Formats:https://www.metaboanalyst.ca/docs/Format.xhtml
なお、下記コマンドはMetaboAnalystで解析し、得た分析コマンドを含みます。分析コマンドを実行するたびに作業フォルダに結果が出力されます。
# ローカルに保存した「cow_diet.csv」データを読み込む
FilePath <- file.choose()
#「MetaboAnalystR」パッケージを読み込む
library("MetaboAnalystR")
# 以下、MetaboAnalystサイトで解析し、得た分析コマンド
# 結果が同一であるかをチェックする
# PID of current job: 1182864
mSet <- InitDataObjects("conc", "stat", FALSE)
mSet <- Read.TextData(mSet, FilePath, "rowu", "disc")
mSet <- SanityCheckData(mSet)
mSet <- ReplaceMin(mSet);
mSet <- PreparePrenormData(mSet)
mSet <- Normalization(mSet, "NULL", "LogNorm", "NULL", ratio=FALSE, ratioNum=20)
mSet <- PlotNormSummary(mSet, "norm_0_", "png", 72, width=NA)
mSet <- PlotSampleNormSummary(mSet, "snorm_0_", "png", 72, width=NA)
mSet <- ANOVA.Anal(mSet, F, 0.05, "fisher")
mSet <- PCA.Anal(mSet)
mSet <- PlotPCAPairSummary(mSet, "pca_pair_0_", "png", 72, width=NA, 5)
mSet <- PlotPCAScree(mSet, "pca_scree_0_", "png", 72, width=NA, 5)
mSet <- PlotPCA2DScore(mSet, "pca_score2d_0_", "png", 72, width=NA, 1,2,0.95,0,0)
mSet <- PlotPCALoading(mSet, "pca_loading_0_", "png", 72, width=NA, 1,2);
mSet <- PlotPCABiplot(mSet, "pca_biplot_0_", "png", 72, width=NA, 1,2)
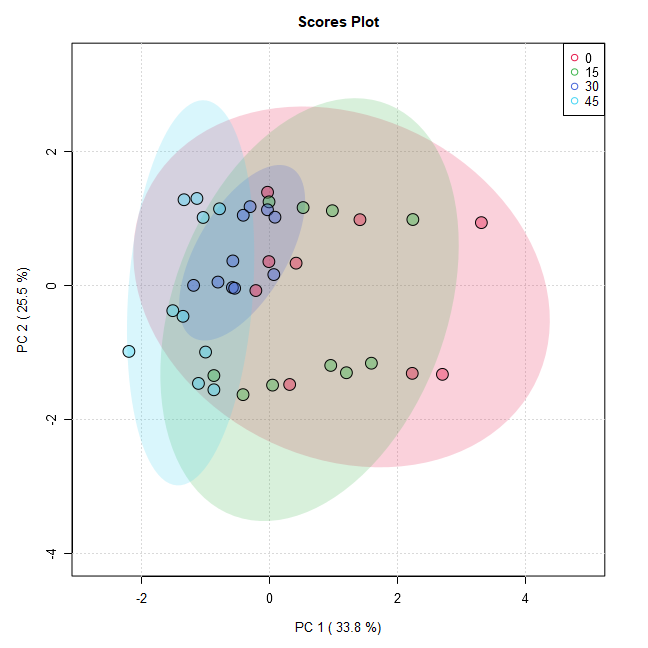
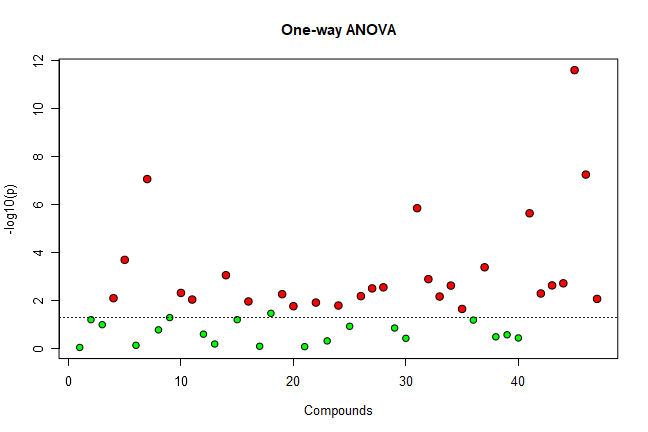
mSet <- PlotPCA3DLoading(mSet, "pca_loading3d_0_", "json", 1,2,3)出力例
・PlotPCA2DScoreコマンド
・ANOVA.Analコマンド
少しでも、あなたの解析が楽になりますように!!