
マイクロアレイで発現変動を示した遺伝子の中からPubMedから入手したエビデンスリストに含まれる遺伝子を抽出するコードを作成しました。ご紹介します。
解析の前準備
- Rの導入についてはこちらから。
- XML, XLConnectパッケージの導入
*パッケージの一括導入は下記コードで可能です。
[code language=”R”]
install.packages(c(“XML”, “XLConnect”))
[/code]
- 発現変動を示した遺伝子名(Official Symbol)が入力されたエクセルファイル(試験結果ファイル)
*遺伝子名(Official Symbol)の他にfold changeやflagの情報が入力されていても問題はありません。必須情報は遺伝子名です。
PubMedのエビデンスリストを入手
遺伝子網羅解析を実施する方はPubMedをご存知かと思います。参考までに、PubMedで遺伝子のエビデンス入手方法を紹介します。
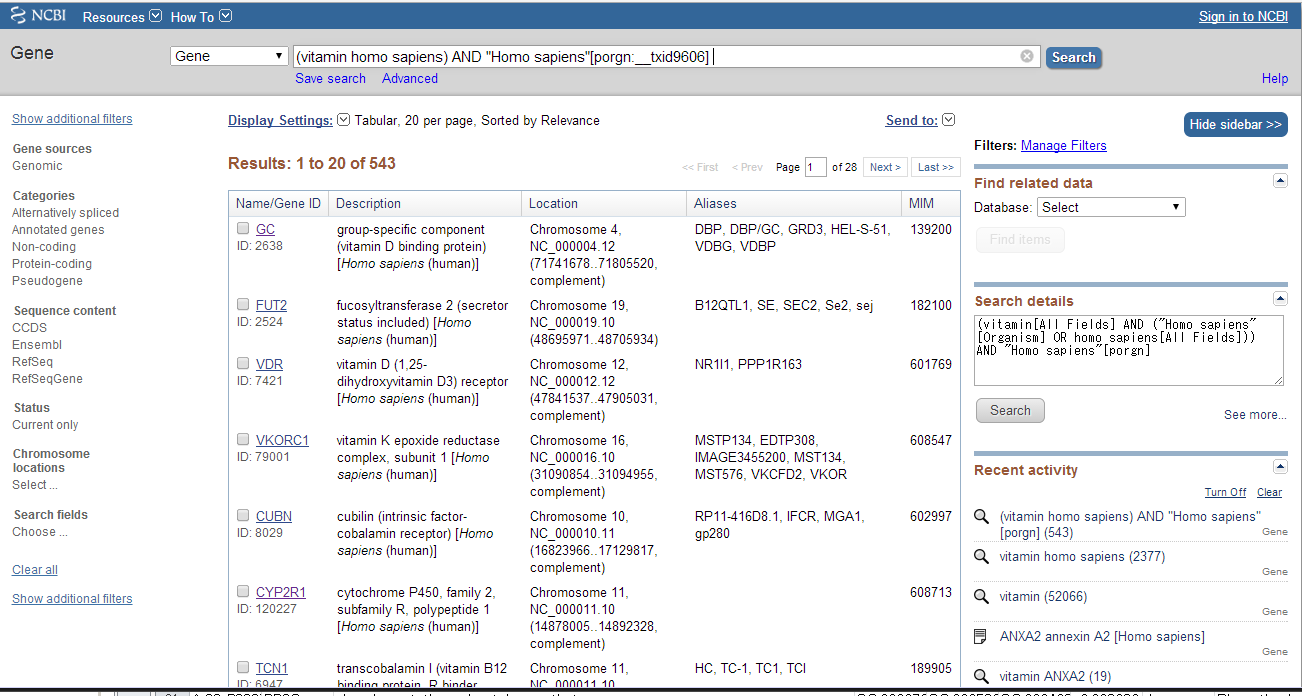
- PubMedにアクセスする。http://www.ncbi.nlm.nih.gov/pubmed
- 検索入力枠左から検索対象のデータベースを「Gene」に変更してエビデンスリストを入手したいキーワードを入力する。検索するとキーワードに関連する遺伝子が検索結果に表示されます。
- 検索結果の右上に表示されるSend toをクリックしFile→FormatをxmlとしCreate Filを押す。そうすると、xmlファイルがダウンロードできます。
*なお、キーワードによってはxmlファイルサイズが大変大きくなりますのでご注意を。
コードの実行
実行すると、試験結果ファイルからエビデンスリストに含まれる遺伝子を抽出し、最終行にPubMedの遺伝子機能のサマリーが追加された「結果.csv」が出力されます。詳細はコードにコメントを記述していますのでご確認ください。
[code language=”R”]
###ライブラリーの読み込み#####
library(tcltk)
library(XML)
library(XLConnect)
########
###解析遺伝子データで遺伝子名列番号を入力#####
AnaGeneName
少しでも、あなたの実験解析が楽になりますように!!
もし、不明点がありましたらこちらからお問い合わせください。

